


В статье собраны самые частые вопросы родителей детей со спинальной мышечной атрофией, связанные с диагностикой, причинами и степенью тяжести заболевания. На вопросы родителей отвечают врач-генетик Наталья Ветрова и врач-невролог Анна Кокорина.
От чего зависит форма заболевания?
От чего зависит форма заболевания. Допустим, у человека нарушения в гене SMN 1. Это в целом понятно, что будет СМА. Но что происходит при той или иной форме в генах? Есть относительно легкие формы СМА, есть тяжелейшие — повреждение в генах одно и то же или разное?
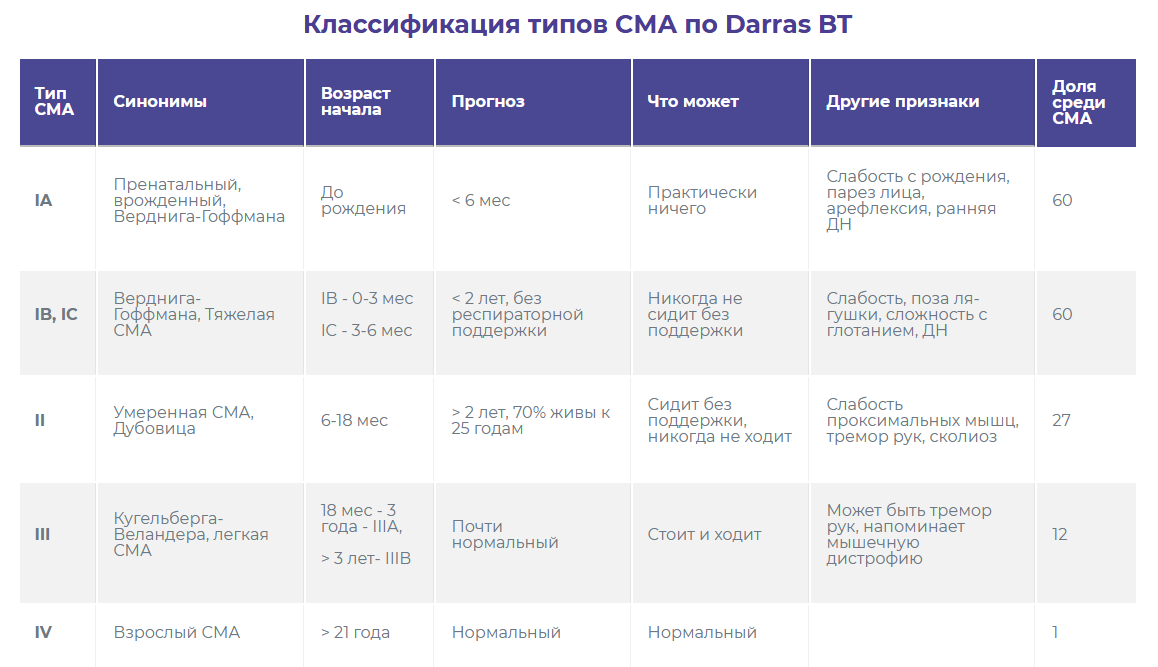
Врач-генетик:Тяжесть течения СМА, обусловленной мутациями в гене SMN1 (которые приводят к полному отсутствию синтеза белка выживаемости мотонейронов), зависит от модифицирующих факторов. Одним из основных факторов, влияющих на возраст начала и тяжесть клинических проявлений является количество копий гена SMN2. Ген SMN2 очень похож на ген SMN1, но продуктом гена SMN2 является нефункциональный белок. Однако, в результате ошибок синтеза белка, которые иногда случаются в нашем организме, с гена SMN2 может синтезироваться небольшое количество правильного, функционального белка, необходимого для работы двигательных нейронов. Именно поэтому даже при полном отсутствии нормального белка с гена SMN1 количество копий гена SMN2 влияет на тяжесть симптомов. Чем больше копий гена SMN2, тем большее количество нужного белка в организме и тем позже и легче протекает заболевание. Однако такая корреляция наблюдается не всегда, предполагаются и изучаются и другие модифицирующие факторы, и пока не все из них нам известны.
Врач-невролог: Действительно, мутация едина для любого клинического типа из проксимальных СМА 5q. В настоящее время считается, что тяжесть клинической картины зависит от модифицирующих (способных изменять течение заболевания) факторов. К основным из них, а также наиболее изученным к настоящему времени генетиками по своей модифицирующей болезнь роли, относится псевдоген SMN2. С него возможен синтез небольшого количества белка выживаемости мотонейронов. Его модифицирующий заболевание потенциал принято оценивать исследованием числа копий. Таким образом, чем больше копий гена SMN2, тем менее агрессивна клиника течения болезни. Важно уточнить, что эта корреляция не абсолютна. Сегодня изучаются и другие модификаторы течения СМА, как эндогенные, в том числе генетические (которые работают внутри генетических процессов, протекающих в организме человека на этапах до производства конечного продукта — белка SMN), так и экзогенные.
При раннем скрининге как-то можно спрогнозировать форму СМА?
Врач-генетик: Пока не все модифицирующие факторы нам известны, при неонатальном скрининге такой прогноз возможен, но пока не однозначен. Основной фактор, на основании которого делает прогноз о форме СМА – это количество копий гена SMN2. Прогнозирование очень важно при принятии решения о назначении досимптоматического лечения, когда у новорожденного выявляется делеция в гене SMN1 в гомозиготной форме. В будущем при внедрении неонатального скрининга на СМА в нашей стране можно будет ориентироваться на опыт других стран. Так, например, опыт проведения такого скрининга в ряде европейских стран и США показал необходимость раннего назначения лечения не только пациентам, имеющим от 0 до 3 копий гена SMN2, но также детям с 4 копиями гена SMN2, для которых ранее применялась выжидательная тактика.
Врач-невролог: При раннем скрининге, когда у ребенка обнаружена мутация, характерная для СМА¸ спрогнозировать клинический тип заболевания и соответственно тяжесть его течения может помочь число копий гена SMN2. При этом важно знать, что здесь может не быть однозначной корреляции. Поэтому в первые месяцы жизни ребенка, при условии уже подтвержденного ранее диагноза СМА, врач-невролог самостоятельно способен прогнозировать тип болезни, полагаясь на исходные клинические данные (симптомы основного заболевания и его осложнения).
Может ли СМА при генетическом подтверждении вообще не проявиться или проявиться в позднем возрасте?
Может ли СМА при генетическом подтверждении вообще не проявиться или проявиться в позднем возрасте? Например, я слышала, что у некоторых признаки СМА проявились после 50 лет. От чего это зависит?
Врач-генетик: Да, существует поздняя форма СМА, или СМА 4 типа, когда у человека симптомы заболевания появляются во взрослом возрасте и при этом сохраняется способность ходить. Одним из основных известных факторов, от которых зависит возраст начала и тяжесть клинических проявлений является количество копий гена SMN2, у таких людей, чаще всего, их от 4 до 6. Кроме того, есть поздние формы СМА, связанные с мутациями в других генах, не в SMN1.
Врач-невролог: Если мы говорим все о том же заболевании, связанном с мутацией в гене SMN1 (проксимальные СМА 5q), то оно представляет собой небольшую группу клинически гетерогенных форм — СМА I-IV. Действительно, клинический тип СМА IV дебютирует во взрослом возрасте и характеризуется относительно благоприятным течением.
Что вызывает спонтанную мутацию, которая приводит к СМА?
Сейчас все чаще говорят не о как таковом наследственном заболевании СМА, а о спонтанной мутации. То есть, произошла внезапная мутация гена у ребёнка, родители которого были носителями. Что, на ваш взгляд, влияет на эту спонтанную мутацию? Плохая экология? Чернобыль? ГМО?
Врач-генетик: Так называемые новые мутации – могут быть индуцированными (следствием физического, химического или инфекционного воздействия), а могут быть спонтанными (случайные изменения в геноме происходят постоянно, в большинстве случаев они не приводят к нарушениям синтеза белка и не являются мутациями, но иногда бывает так, что изменение касается какого-то очень важного участка генома и тогда оно может повлиять на белок). Причиной СМА, обусловленной мутациями в гене SMN1, в подавляющем большинстве случаев являются унаследованные мутации. И в таком случае мутации действительно наследуются от родителей-носителей, которые всегда здоровы и не подозревают, что несут в своем геноме «скрытую» мутацию. Об это нельзя никак узнать, если заранее не провести специальный тест на носительство.
Варианты мутаций de novo или новых мутаций, обусловливающих СМА, возможны но редки. В таком случае обычно только один из родителей является носителей мутации в гене SMN1, а у второго мутация формируется случайно, в его половой клетке.
Врач-невролог: СМА — это наследственное заболевание, когда мутация в гене является унаследованной от родителей-носителей/больных. Новые мутации в происхождении СМА крайне редки.
Чем отличается СМА, вызванное нарушением в других генах, от классической формы СМА?
Генетический анализ на СМА отрицательный, но внешне человек похож на больного СМА, может ли быть мутация в генах, которая вызывает СМА, но находится она не в обычном гене? Если не выявили нарушений в гене SMN1, но по клиническим признакам ставят СМА, то в каких генах может быть нарушение? Чем отличается СМА, вызванное нарушением в других генах, от классической формы СМА? Какое течение заболевания при других, редких формах СМА.
Врач-генетик: Для большинства наследственных нервно-мышечных заболеваний характерен такой феномен, как генетическая гетерогенность. Это означает, что клинические проявления заболевания и результаты инструментальных исследований могут быть одинаковыми, но генетическая причина их может быть разной. Например, клиническая картина заболевания спинальной мышечной атрофии может быть обусловлена мутациями в нескольких разных генах, при этом у всех пациентов будет повреждение мотонейронов передних рогов спинного мозга. А вот возраст начала, тяжесть и тип наследования могут быть разные. Просто наиболее частой причиной СМА являются мутации в гене SMN1 (это связано с высокой частотой бессимптомного носительства этой мутации у здоровых людей), поэтому всегда поиск причины СМА начинают с анализа делеций гена SMN1. Если их не находят, тогда поиск причины продолжают. Течение заболевания будет зависит от конкретного гена, в котором найдена поломка. Есть крайне тяжелые летальные формы СМА, а есть очень мягкие поздно возникающие формы.
Врач-невролог: Говоря о СМА в целом (о группе СМА, без прицела на СМА 5q), необходимо сказать о генетической гетерогенности этого явления (спинальной мышечной атрофии). То есть причиной СМА может стать генетическая поломка в разных генах (кроме гена SMN1, расположенного на пятой хромосоме), некоторые из них сегодня остаются не изученными и не картированными. В таком случае мы будем говорить о СМА, как о синдроме, который является ядром клинической картины развившейся у пациента болезни, что подтвердят объективный неврологический статус пациента, проведенные инструментальные исследования (например, ЭМГ).
Электромиография (ЭМГ) – это диагностический метод, посредством которого специалисты оценивают функциональное состояние скелетных мышц и окончаний периферических нервов.
Среди всех генетически гетерогенных СМА существуют относительно благоприятные (доброкачественные) формы и тяжелейшие формы. Есть формы, сочетающие синдром спинальной мышечной атрофии и другие расстройства (патологию (гипоплазию) в отделах головного мозга, респираторные нарушения — РДС, артрогрипоз и другое). Отличать от СМА 5q их будут особенности клинической картины, иное распределение мышц, вовлеченных в процесс атрофии, дополнительные симптомы, которых нет при СМА 5q, а так же тип наследования. СМА, развитие которой связано с мутацией в гене SMN1, расположенном на 5q13, принято наименовать проксимальной СМА 5q.
Чем отличается течение СМА при повреждении в гене SMN1 и СМА с преимущественным поражением ног?
Чем отличается течение СМА при повреждении в гене SMN1 и СМА с преимущественным поражением ног, обусловленной мутациями в гене DYNC1H1 с аутосомно-доминантным типом наследования. Различается ли поддерживающая терапия?
Врач-генетик: Заболевания отличаются по характеру течения и сопутствующим симптомам. Характер течения при СМА, обусловленной мутациями в гене DYNC1H1, не прогрессирующий или медленно прогрессирующий, возможен когнитивный дефицит. Симптоматическая поддерживающая терапия не отличается от таковой при терапии СМА, обусловленной мутациями в гене SMN1.
Врач-невролог: Здесь важно сказать, что СМА, обусловленная мутациями в гене DYNC1H1, изучена и описана гораздо меньше, нежели проксимальная СМА 5q. Известно, что ген DYNC1H1 способен подвергаться множеству мутаций, из них идентифицировано более 60. Каждая из этих мутаций или их сочетание способно приводить к развитию различных по клиническим проявлениям заболеваний, так одним из наиболее частых как раз является СМА с преимущественным поражением мышц нижних конечностей. В целом заболевание характеризуется аутосомно-доминантным типом наследования гетерозиготной мутации (в семье встречается довольно часто, например, есть у родителя и у ребенка), широким возрастным диапазоном дебюта, поражением проксимальных и дистальных мышц нижних конечностей, деформациями стоп и контрактурами суставов нижних конечностей (в первую очередь голеностопных), нарушением походки, неспособностью к сложным двигательным актам (бег/прыжки), развитием компенсаторного лордоза поясничного отдела позвоночника. При более «типичном» варианте (если такое выражение позволительно при малочисленных описанных случаях) чаще наблюдается благоприятное течение с длительной сохранностью самостоятельной ходьбы. Есть формы, которые развивают более тяжелые и распространенные контрактуры суставов нижних конечностей, тогда ходьба становится затруднительной. Иногда форма СМА с преимущественным поражением мышц нижних конечностей сочетается с интеллектуальными нарушениями (умственной отсталостью). Что же касается симптоматической терапии, то она принципиально ничем не отличается от таковой при проксимальных СМА 5q.
Могут ли тяжёлые роды спровоцировать более сложную форму СМА?
Могут ли тяжёлые роды, при которых ребёнок получил повреждения, спровоцировать более сложную форму СМА? Возможно ли, что в определённых случаях при поврежденном гене SMN1 ребёнок был бы просто носителем, а тяжёлые роды спровоцировали активную форму СМА?
Врач-генетик: Если ребенок был носителем мутации в гене SMN1 в гетерозиготной форме, он и после тяжелых родов останется носителем и СМА у него не разовьется. Но если у ребенка мутация в гомозиготной форме – тогда у такого ребенка будет 2 заболевания – СМА и перинатальное поражение ЦНС, конечно в таком случае состояние ребенка будет хуже, чем у детей с одним диагнозом.
Врач-невролог: Нет, не могут. Носитель останется носителем, а больной — больным. Тяжелые роды способны оказать неблагоприятный фон для новорожденного, больного СМА, вероятно, могут спровоцировать более ранний клинический дебют болезни.
Влияет ли внутриутробная гипоксия, «страдания плода» во время беременности на проявление СМА или усложнение формы заболевания?
Врач-генетик: Наличие любой тяжелой сопутствующей патологии может влиять на течение СМА.
Врач-невролог: Любой отягощенный фон при СМА способен влиять на течение болезни.
Связаны ли СМА и спинальная травма во время родов?
Часто задают вопрос: во время родов у новорожденного произошла травма спинного мозга. Через некоторое время у него диагностируют СМА, оно, как известно, заключается не только в нарушении функции гена, но и в повреждении передних рогов спинного мозга. Это может быть как-то взаимосвязано?
Врач-генетик: Нет. Родовая травма может приводить к повреждению спинного мозга – механическому или сосудистого характера, при этом белок выживаемости мотонейронов будет синтезироваться и будет потенциал для восстановления функции передних родов спинного мозга. СМА – это следствие мутации в гене, характер повреждения рогов спинного мозга при этом обусловлен отсутствием конкретного белка.
Врач-невролог: Причиной СМА никогда не может быть травма спинного мозга, только генетическая мутация. И мотонейроны передних рогов спинного мозга страдают и погибают как раз из-за генетической мутации, следствием которой является отсутствие жизненно необходимого им белка «выживаемости мотонейронов». У ребенка со СМА, как и у любого другого, теоретически возможно повреждение спинного мозга (либо его корешковых структур) во время продвижения по родовым путям матери. В таком случае возможно развитие ранней (острой) клиники повреждения спинного мозга или его структур. Эти симптомы будут опережать симптомы развития СМА (в случае, если речь не идет о врожденной тяжелейшей форме СМА 0). Обязательно при травме можно найти уровень повреждения спинного мозга с помощью дополнительных инструментальных методов исследования. Не нужно искать взаимосвязь между этими двумя состояниями.
Если при обследовании обнаруживается, что существуют нарушения в гене, отвечающем за наличие СМА. Заболевание обязательно проявится или существует некий «спящий» режим?
Врач-генетик: 99,9% рано или поздно проявится. «Спящий» режим бывает при наследственных заболеваниях, но мутации в гене SMN1 практически 100% проявятся.
Врач-невролог: Да, заболевание проявится.
Может ли быть ошибка в результате анализа? Имеет ли смысл пересдать анализ в другой лаборатории?
Врач-генетик: Референсным методом выявления делеции в гене SMN1 является MLPA. Если ранее был использован другой метод и есть сомнения в диагнозе – можно пересдать.
Врач-невролог: Теоретически — да, может быть. Если клинические проявления болезни расходятся с генетическим диагнозом, — смысл пересдачи анализа возрастает.
Нужно ли сдавать анализ на количество копий гена SMN2 и зачем?
Врач-генетик: Количество копий гена SMN2 имеет значение у пациентов с гомозиготной делеций в гене SMN1 для уточнения типа СМА, прогноза и возможного лечения. Также может влиять на попадание в ту или иную группу клинических испытаний.
Врач-невролог: Мы уже говорили, что псевдоген SMN2 — основной изученный модификатор (способный изменять течение болезни) СМА 5q. Поэтому для того, чтобы знать свое заболевание «в лицо», лучше сдавать такой анализ. Надо сказать, что этот показатель рассматривался в качестве критерия для участия в клинических испытаниях и программах расширенного доступа препаратов. Он также остается дополнительным критерием для генной терапии СМА.
Количество копий гена SMN2 влияет на выбор терапии и средств реабилитации?
Врач-невролог: На этот выбор первоначально влияет состояние пациента. Однако, если говорить отдельно о лечении (я сейчас имею в виду этиопатогенетическую генную терапию), то в некоторых странах в показания к назначению препарата включено число копий гена SMN2 (не более 3 копий).
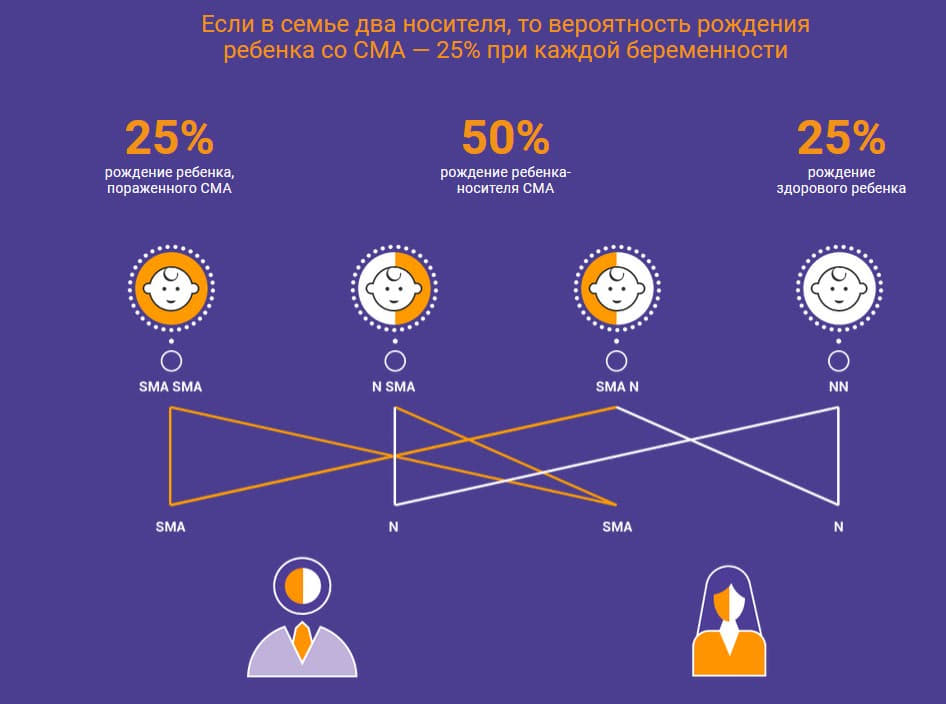
Если вероятность рождения ребенка со СМА у двух носителей равна 25 процентам, значит ли это, что при рождении ребенка со СМА следующие трое у этих родителей не будут больны?
Врач-генетик: Нет. Может быть так, что все 3 будут со СМА. На таких маленьких выборках, как число детей у человека такие статистические законы не действуют. Например, если бы у человека было потомство, составляющее 100 детей – то в семье носителей мутации в гене SMN1 было бы приблизительно 25% детей со СМА, 50% носителей и 25% неносителей мутации в гене SMN1. Но это не было бы обязательно 25 детей со СМА и 75 детей здоровых, это все вероятностные события, здесь нельзя однозначно прогнозировать.
Врач-невролог: Нет, совсем не значит. У двух супругов-носителей могут родиться все дети с заболеванием СМА, могут родиться все носителями, а так же все здоровыми. Возможны любые вариации в процентном соотношении. Закон расщепления признаков действует в математической плоскости и не может быть показательным на примере потомства одной семьи.
У нас в семье ни у кого не было СМА, почему же заболел мой ребенок?
Врач-генетик: СМА – является рецессивным заболеванием. В таких случаях больной ребенок рождается у абсолютно здоровых людей, в подавляющем большинстве случаев с «чистой» родословной. Рецессивные заболевания проявляются, когда ребенок получает копии гена с мутацией от обоих родителей. При этом родители носят мутации бессимптомно, в скрытом виде. Каждый человек является носителем подобных скрытых мутаций в своем геноме и об этом можно узнать только проведя специальное генетическое тестирование на этапе планирования беременности.
Врач-невролог: Это как раз «типичный» случай для СМА. СМА имеет аутосомно-рецессивный тип наследования. Это значит, что заболевание проявится в том случае, когда ребенок унаследует мутацию сразу от обоих, не подозревающих об этом, родителей-носителей. Таким образом, для рождения больного ребенка должны встретиться и создать семью два носителя это рецессивной мутации. Подтверждается это и тогда, когда у этих родителей рождаются несколько больных СМА детей. Рецессивные генетические заболевания в целом характеризуются тем, что встречаются достаточно редко и могут вообще не присутствовать в родословной родителя.
Мы планируем завести еще детей, но не хотим, чтобы у них было СМА. Какие меры можно предпринять, чтобы родить здорового ребенка?
Врач-генетик: Можно провести пренатальную диагностику – в сроке беременности 10-11 нед через прокол живота получить материал плода для исследования и проверить у него наличие мутаций в гене SMN1. При подтверждении делеции в гомозиготной форме – предлагается прерывание беременности. Также возможно тестирование эмбрионов, полученных методом ЭКО, – на мутации в гене SMN1. В таких случаях эмбрион с гомозиготной делецией просто не переносится женщине.
Врач-невролог: Для этого предусмотрено медико-генетическое консультирование, предлагаются к рассмотрению варианты пренатальной и предимплантационной диагностики с применением репродуктивных технологий во втором случае.
Если человек болен СМА, значит ли это, что его ребенок тоже будет болен СМА?
Врач-генетик: Нет. Это будет зависеть от генетического статуса супруга/и. Если супруг не является носителем мутации в гене SMN1 – ребенок будет 100% здоровым носителем.
Врач-невролог: Нет, не значит. В этом случае всё будет зависеть от второго супруга, — от того, носит ли он мутацию
Если у родного брата или сестры СМА, какие шансы родить ребёнка со СМА? Изменяется ли ситуация, если это двоюродный или троюродный родственник?
Врач-генетик: Ситуация всегда зависит от генетического статуса обоих супругов, планирующих беременность. Если в семье есть любой родственник со СМА – необходимо провести исследование на носительство делеции в гене SMN1 (или другой тест в зависимости от причины СМА у родственника), и если обнаруживается мутация – сделать соответствующий тест супругу/е.
Воач-невролог: Если в семье/родословной (при условии кровного родства) есть больные СМА, то риск рождения больного ребенка существует. Однако трудно утверждать, что он превышает общепопуляционный риск. В этом случае решающее значение имеет генетический статус, как родственника пациента со СМА, который планирует создавать потомство (то есть человека, который задает нам этот вопрос), так и его супруга(и); а так же то, как сработает закон наследования (в случае, если вдруг гипотетические родители оба окажутся носителями мутации).
Взрослый человек со СМА решил завести ребёнка, партнёр по анализу носитель СМА, какие риски рождения больного ребёнка? А если партнер не носитель гена СМА?
Врач-генетик: Если один из супругов является носителем делеции гена SMN1 в гомозиготной форме (болен СМА), а другой является бессимптомным носителем делеции в гетерозиготной форме — риск рождения ребенка со СМА — 50%. Если один из супругов является носителем делеции гена SMN1 в гомо- или гетерозиготной форме, а другой нет, риск рождения ребенка со СМА низкий.
Врач-невролог: Говоря о реализации генетических законов «на деле», мы часто можем рассуждать только в рамках некой математической модели (не в абсолютных числах по количеству больных, прогнозируемых к рождению в конкретной семье). Поэтому, отвечая непосредственно на вопрос, — если один родитель болен СМА, а другой является здоровым и не носит мутацию, то все дети этих родителей будут клинически здоровыми носителями данной мутации; если же второй родитель будет носителем мутации, тогда риск рождения больного ребенка 50/50 (то есть высокий).
Если ребенку со СМА пересадить какой-нибудь орган, он поправится?
Врач-генетик: Суть заболевания заключается в генетической поломке, которая ведет к тому, что в организме отсутствует белок выживаемости мотонейронов и поэтому перестает работать спинной мозг. Ученые пытаются воздействовать на суть и заставить ген синтезировать нужный для организма белок.
Врач-невролог: Пересадка никакого органа, в случае со СМА, никак не поменяет суть и течение этого заболевания.
Влияют ли прививки на возникновение СМА, ее форму и тип?
Врач-генетик: Нет, не влияют. Возникновение и тип СМА обусловлены генетическими мутациями.
Врач-невролог: Нет, не влияют.
Данный материал носит исключительно информационный характер и не может служить рекламой. Рекомендации относительно индивидуального применения любого лекарственного препарата или назначения лечения следует получать у вашего лечащего врача.