


Спинальная мышечная атрофия – это заболевание, при котором повреждается ген SMN1. В результате мутации у человека не вырабатывается белок, который нужен для выживания мотонейронов. Мотонейроны — важная часть спинного мозга человека, от них по длинным нервам идут сигналы к скелетным мышцам организма. Если мотонейрон получает недостаточно белка для своего питания, то он быстро устает. Ему приходится работать и за себя, и «за того парня». Многие мотонейроны просто гибнут от «усталости», — нагрузка на оставшиеся возрастает, и в результате они работают с чудовищной перегрузкой. От усталости они не могут посылать сигналы к скелетным мышцам, благодаря которым человек ходит, сидит, лежит, глотает или дышит. Мышцы остаются без работы, потому что их никто ничего не просит сделать, и постепенно отмирают.
Дефицит важного белка, который не вырабатывает ген SMN1, приводит к атрофии (потере жизнеспособности и уменьшению размера) мышц, и в результате человек постепенно теряет способность ходить, удерживать и управлять собственным телом, самостоятельно сидеть, есть, глотать и дышать. Упрощенный механизм СМА можно увидеть на схеме.

К счастью, у человека есть еще ген SMN2, который дублирует SMN1. Он производит нефункциональный белок и небольшое количество нормального белка, который позволяет мотонейронам человека выживать на очень голодном пайке. В результате человек со СМА сохраняет какую то часть своих двигательных, глотательных и дыхательных функций.
От чего зависит тип СМА и тяжесть болезни?
Если максимально упростить картину, то у человека может быть разное число копий гена SMN2. Теоретически, чем больше у пациента со СМА таких копий, которые вырабатывают маленькое количество нужного мотонейронам белка, тем легче будет протекать заболевание, человек сохранит больше жизненно важных функций и дольше проживет. На практике есть слишком много нюансов, чтобы напрямую связывать тяжесть болезней с числом копий SMN2.
При этом у людей признаки СМА появляются в разное время: от первых дней жизни до взрослого возраста.
Существует четыре типа спинальной мышечной атрофии. Чем раньше начинается заболевание, тем тяжелее оно протекает. В 95 % случаев причиной СМА становится делеция (выпадение) участка генетического кода.
По международным стандартам типы СМА различаются несколькими важнейшими признаками:

Болезнь Верднига-Гофманна (СМА 1), возраст дебюта заболевания от 0 до 6 месяцев, максимальная (двигательная) функция у таких детей: сами не могут сидеть. При естественном течении заболевания 92 % этих деток погибает до возраста двух лет.

Болезнь Дубовица (СМА 2). Дебют заболевания происходит с 6 до 18 месяцев. Такие пациенты никогда не могут самостоятельно стоять, они могут самостоятельно сидеть. Продолжительность их жизни может быть больше двух лет

Болезнь Кугельберга-Велендер (СМА 3). Дебют заболевания происходит позже 18 месяцев, такие пациенты развиваются и нормально ходят, но в какой-то момент заболевание начинает прогрессировать. Они перестают самостоятельно передвигаться и садятся. Хотя продолжительность жизни у них тоже сильно ограничена, но они могут прожить несколько десятилетий.

Четвертый тип мы почти не видим в детской клинике. Такие пациенты дебютируют в подростковом и взрослом возрасте, они могут потерять способность к самостоятельному передвижению. Продолжительность жизни — десятилетия.
|
Тип СМА |
Возраст дебюта заболевания |
Максимальная функция |
Средняя продолжитель-ность жизни |
Типичные проявления |
|
Тип 1 Болезнь Верднига-Гофманна |
0-6 месяцев |
Не сидит |
< 2-х лет |
Глубокая слабость и гипотония, трудности контроля головы, слабый крик и кашель, трудности с глотанием и выведением мокроты, осложненное течение ОРВИ, развитие дыхательной недостаточности, высокие риски аспирационной пневмонии, очень высокая скорость прогрессирования заболевания |
|
Тип 2 Болезнь Дубовица |
6-18 месяцев |
Не стоит |
> 2-х лет |
Задержка моторного развития и набора веса, слабый кашель, тремор рук, контрактуры, сколиоз и деформация грудной клетки. |
|
Тип 3 Болезнь Кугельберга-Веландер |
Старше 18 месяцев |
Стоит и ходит |
Десятилетия |
Мышечная слабость различной степени выраженности, крампи (судороги в икроножных мышцах), контрактуры и гипермобильность суставов, потеря способности ходить по мере прогрессирования заболевания |
|
Тип 4 Поздний тип |
В подростковом или взрослом возрасте |
Могут потерять способность к самостоятельному передвижению |
Десятилетия |
Прогрессирующая проксимальная мышечная слабость, снижение сухожильных рефлексов, фасцикуляции |
Чуть более подробно и наглядно об особенностях разных типов СМА можно посмотреть по ссылке
Почему рождается ребенок со СМА?
Спинальная мышечная атрофия — это генетическое заболевание. Каждый сороковой житель планеты — здоровый носитель СМА. Чтобы в семье родился ребенок со СМА, нужно, чтобы такими носителями были оба родителя. В этом случае в 25% случаях ребенок родится здоровым, в 50% случаях он получает мутацию гена SMN1 от папы или от мамы и тоже становится просто носителем, а еще в 25% случаях он наследует мутацию гена SMN1 от обоих родителей и рождается с этой болезнью.
Отметим, что у здоровых носителей мутации гена SMN1 есть один полноценный ген и один ген с мутацией. Наличие одного полноценного гена достаточно для выработки необходимого количества нормального белка и правильного функционирования мотонейронов. Так что носитель от человека без поломки этого гена отличается лишь потенциальным риском рождения ребенка со СМА.
Более наглядно это показано на схеме
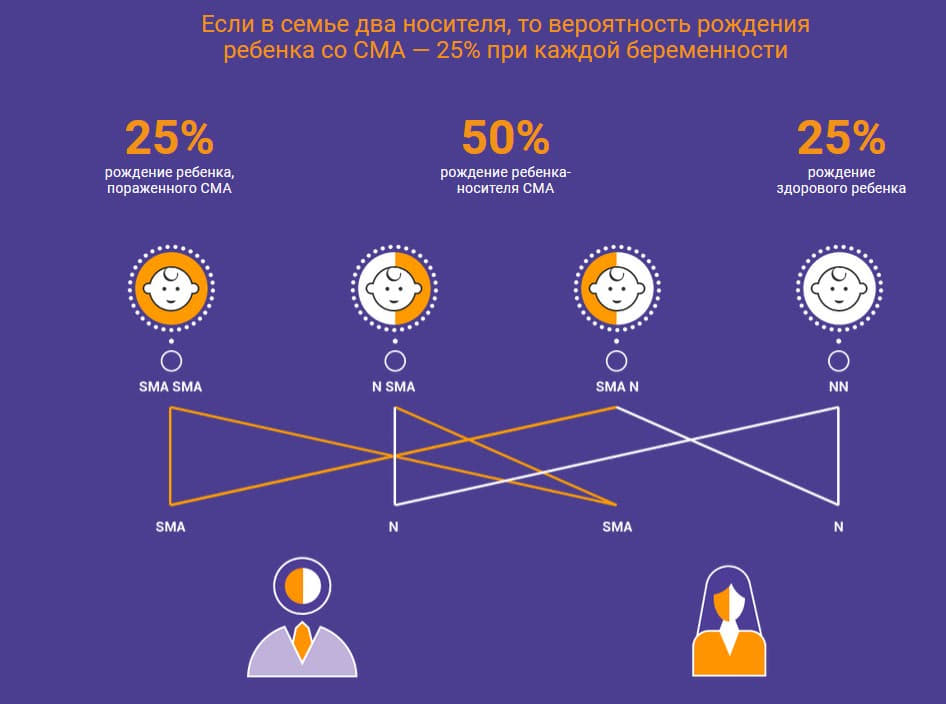
К сожалению, в реальности генетические особенности человека срабатывают не по математическому принципу.
Кандидат медицинских наук, ведущий врач-генетик ООО «Центра Генетики и Репродуктивной Медицины «Генетико» Наталья Ветрова описывает реальную практику появления детей со СМА в семьях, где есть два носителя:
«В семье могут быть все здоровыми, могут быть все носителями, могут быть все больными, могут через один. Закон 25-50-25 действует на очень больших числах. В реальных семьях люди с мутацией гена SMN1 не могут иметь такое большое потомство, чтобы было распределение так, как у нас на картинке».
Кто и как ставит диагноз "Спинальная мышечная атрофия"
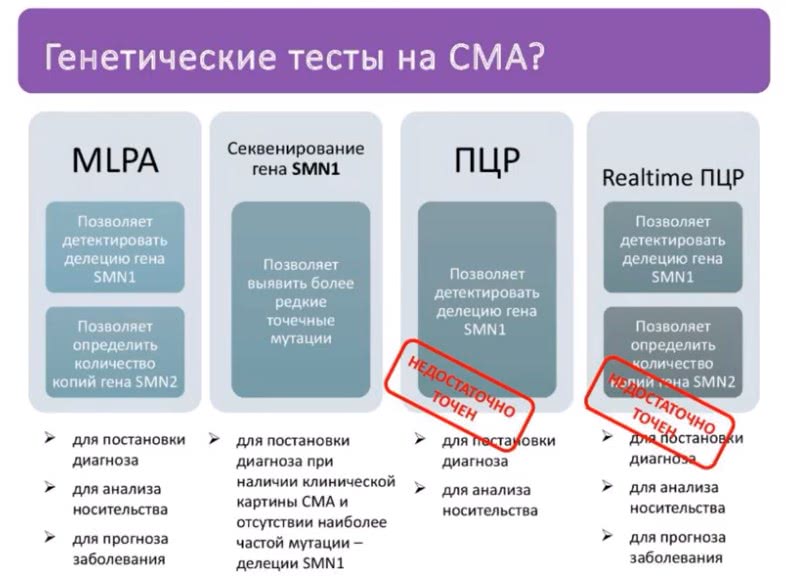
Поставить клинический диагноз СМА врач может после неврологического осмотра пациента с проверкой рефлексов и мышечного тонуса. Еще нужно провести внешний осмотр пациента и сделать биохимический тест.
После этого человек должен пройти еще несколько тестов. Генетическое исследование должно подтвердить или опровергнуть первоначальный клинический диагноз.
Тесты также нужны для уточнения прогноза заболевания и уточнения носительства мутаций гена SMN1 у родителей ребенка.
Подробнее о назначении каждого теста можно узнать, посмотрев на изображение.
Лучший тест на сегодня — MLPA. Он подтверждает причину СМА в 95 % случаев. Для оставшихся 5 % нужно тест секвенирование гена SMN1 (он нужен, если первый тест не подтвердил диагноз, а у пациента есть клинические проявления СМА).
Еще один тест, который может назначить врач, это панель «Нервно-мышечные заболевания» (НМЗ). Он нужен в случае, когда ни MPLA ни секвенирование не смогли найти причину клинических проявлений, которые позволяют врачу заподозрить СМА или другое нервно — мышечное заболевание.
СМА и беременность
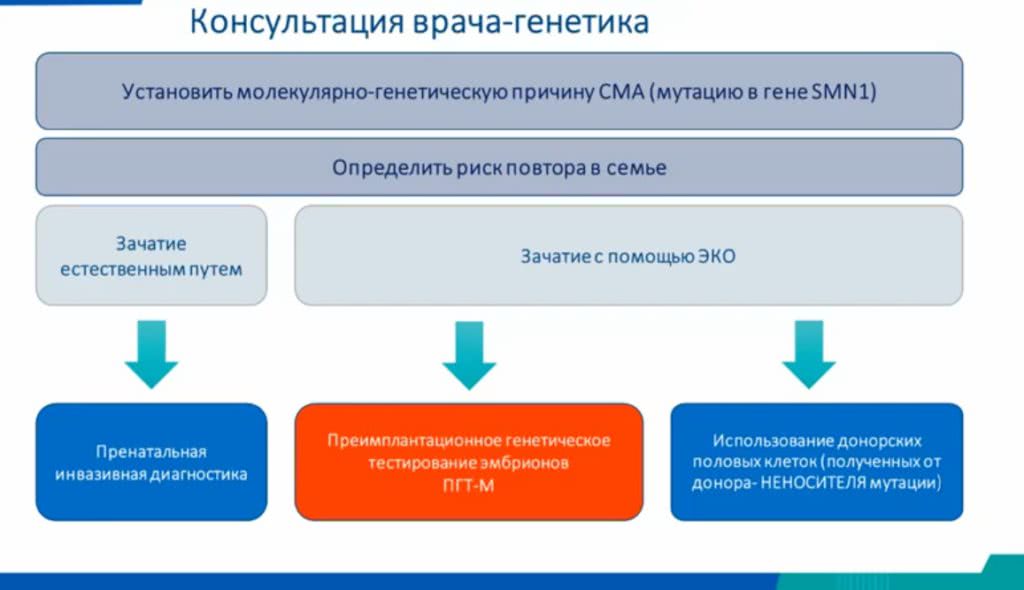
Если в семье уже есть ребенок со СМА или один из будущих родителей — носитель или пациент со СМА, необходимо заранее провести несколько тестов .
Если два основных генетических исследования (MPLA и секвенирование) показывают, что второй родитель тоже носитель мутации гена SMN1, нужно решить, каким образом запланировать и провести беременность, чтобы минимизировать риск появления ребенка со СМА.
Если супружеская пара выбирает естественный путь для зачатия, то она должна пройти пренатальную инвазивную диагностику после десятой недели беременности. Для этого прокалывают живот и делают забор материала у плода. В современных условиях процедура достаточно безопасна и для будущей мамы, и для плода. Существует минимальный риск выкидыша, но он гораздо ниже, чем вероятность рождения ребенка со СМА.
Если супруги предпочитают ЭКО, то они могут пойти двумя путями:
или из нескольких зародышей отобрать здорового, или воспользоваться услугами донора — неносителя СМА. В этом случае донора нужно дополнительно исследовать на наличие мутации в гене SMN1.
При отборе эмбрионов риск возможного повреждения каждого эмбриона стремится к нулю. Отобранный здоровый эмбрион замораживают и подсаживают в матку.
Более подробно о беременности при СМА можно узнать, посмотрев запись вебинара Натальи Ветровой.
Патогенетическая терапия при СМА
В настоящее время в мире есть два уже одобренных препарата для лечения СМА — Нусинерсен (Спинраза) и AVXS -101 (Золгенсма). Ожидается, что в ближайшее время на рынке появится третий препарат RG7916 (Рисдиплам).
Нусинерсен предназначен для пожизненного приема, в случае с Золгенсма речь идет об однократной дозе, но оба этих препарата работают неодинаково. Один решает проблему мышечной слабости, другой — препарат, который модифицирует генные клетки в целом.
Главная задача этих препаратов — восполнить дефицит белка из-за не работающего гена SMN1. О проведенных клинических испытаниях препаратов и о последних новостях лекарственной терапии при СМА можно прочитать в разделе Клинические исследования и лекарства.
Краткий обзор о принципах работы патогенетической терапии при СМА можно прочитать по ссылке.
Эта статья для медицинских работников. Мы не рекомендуем пациентам использовать данные из нее самостоятельно. За консультациями необходимо обратится к врачу. Информация, предоставленная в статье, может относиться к лекарственным препаратам, отпускаемым по рецепту.